
The Cell Death Signalling Lab
Within the Cell Death Signalling Lab run by division head Andreas Villunger, we currently explore the cross talk of the cell death and the cell cycle machinery. Thereby, we focus on understanding signaling events that define thresholds for cell death or survival after mitotic errors. Two aspects keep us busy. How are mitotic errors communicated to the BCL2 family network that controls mitochondrial apoptosis and how is p53, a major tumor suppressor, engaged in this context. Within these studies, we have recently shown that mitotic arrest drives BAX/BAK-mediated cell death by degradation of anti-apoptotic MCL1 and increased abundance of pro-apopotic BIM and NOXA (Figure 1). Following up this finding, we now interrogate who abundance of these proteins is controlled in mitotic cells.
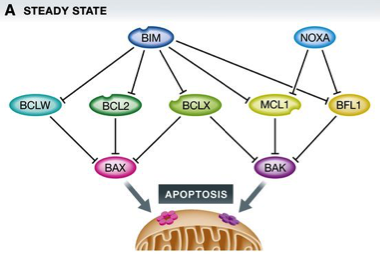
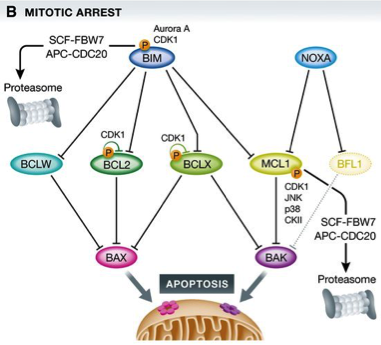
Figure 1: BCL2 family proteins control cell death during mitotic arrest
(A) In healthy cells, a homeostatic equilibrium between cell death initiating BH3‐only proteins (blue), anti‐apoptotic BCL2 family proteins (green), and cell death executioners (pink/purple) is maintained.
(B) Upon perturbation of this equilibrium, for example, by the action of anti‐mitotic drugs and prolonged arrest in mitosis, a series of events, including phosphorylation on and proteasomal degradation of pro‐survival BCL2 proteins, shifts the balance, favoring BAX/BAK1 activation.
Manuel Haschka et al. EMBO Rep. 2018;19:e45440
Moreover, we investigate the role of a multi-protein signaling complex, dubbed the PIDDosome that drives p53 activation upon cytokinesis failure (Figure 2), a process that can prime cells for transformation but may also be the desired outcome during normal organ development, as exemplified in the liver or heart. If you are interested in these topics, please don't hesitate to contact the PI, Andreas Villunger.
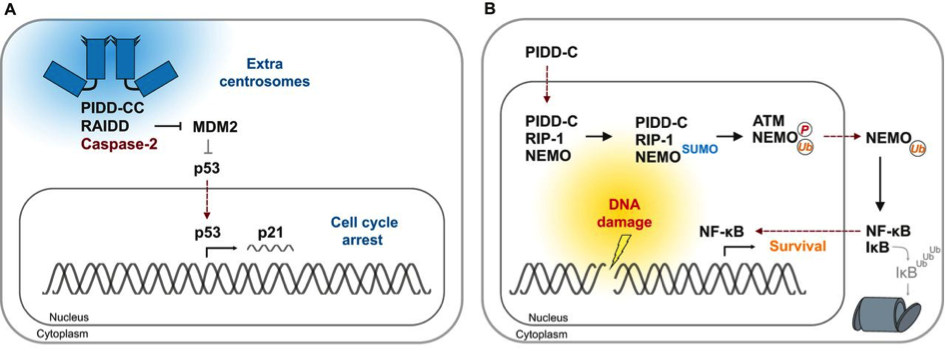
Figure 2: Cues for the formation of distinct PIDDosome signalling complexes
(A) Cytokinesis failure causes accumulation of extra centrosomes (blue). PIDD1 localises to the mature centriole within the centrosome that is characterised by appendage proteins (triangles). The presence of more than one mature centriole causes formation of the Caspase-2−PIDDosome and CASP2 activation. Subsequent MDM2 cleavage by CASP2 generates an N-terminal MDM2-fragment that binds to and stabilises p53. This results in p21-dependent cell cycle arrest.
(B) In response to genotoxic stress that causes double-strand breaks, PIDD-C localises to the nucleus. There, it interacts with RIP-1 to form the NEMO−PIDDosome, thereby facilitating NEMO sumoylation that is, in turn, required for its phosphorylation by ATM and its subsequent mono-ubiquitylation. The latter acts as the nuclear export signal for NEMO. In the cytoplasm, mono-ubiquitylated NEMO targets IκB for proteasomal (grey barrel) degradation, which results in NF-κB activation and transcription of survival and inflammatory genes.
Valentina Sladky et al. J Cell Sci 2017;130:3779-3787